While docking and virtual screening have demonstrated value in drug discovery, they have notable shortcomings. We enumerated these limitations in a previous blog post, but they can be summarized as:
- Knowing your target protein is essential to understand how you can intervene in the disease
- Docking scores and poses are often inaccurate and propagate errors that lead drug hunters astray
- Maximizing search space increases success but is complex and computationally intensive
- Multiparameter optimization is critical for filtering hits but difficult and rarely feasible upfront
We were frustrated by these challenges and decided: it doesn’t have to be this way. So we set out to build the tools we wanted to use in drug discovery. In this post, we’re excited to unveil the docking and virtual screening tools we’ve been building at Deep Origin: BiosimDock and BiosimVS. While we can’t talk about the algorithmic details of our proprietary models, we think it’s important to demonstrate how they perform compared to other tools, allowing potential customers and partners to evaluate them effectively. So let’s dive in!
Access our docking via Balto, our AI assistant. Get a one-month free trial here.
We outperform other models on accuracy of binding affinity and binding pose prediction
Docking and virtual screening tools are meaningful only if they are able to make fast, accurate, and useful predictions. A good model can filter true binders from a broad pool of potential molecules, including false positives that may be similar in chemical properties. In the hit identification stage of drug discovery it can give drug hunting teams a chemically-diverse set of potential hit molecules to evaluate with experimental assays or further computational analysis, helping them narrow the path to a lead candidate. In contrast, a bad model returns many false positives, costing a team money and effort - and potentially leading drug hunters off-course for months or years.
To benchmark, we tested the BiosimDock model on the PDBbind core dataset and the DEKOIS 2.0 dataset.[1], [2], [3], [4] The PDBbind core dataset contains 285 experimental structures of protein-ligand bound complexes across different protein classes. It remains a standard due to widespread use in benchmarking, facilitating comparison between models. It also enables accuracy prediction based on binding poses. DEKOIS 2.0 (Demanding Evaluation Kits for Objective In silico Screening) is an extensively-curated dataset of 81 targets across protein classes, including proteases, kinases, transferases, oxido-reductases, nuclear receptors, and hydrolases. Each target has an accompanying library of true binders and decoys, which have similar physical and chemical properties but do not interact with the target protein. This enables rigorous benchmarking of models for enrichment of true binders over false positives.[4], [5], [6]
Before we dive into results, a warning: benchmarking docking and machine learning models can be tricky due to ‘model memorization’. Often models score highly because similar or identical examples appear in both the benchmarking and training datasets, allowing the model to repeat back the result it observed in the training dataset. These models will score substantially worse when a different benchmark dataset is selected. For example DiffDock, a well known ML-based docking model that has 38% reported accuracy has only 15% accuracy when the test examples are filtered by 30% sequence similarity relative to the PDBbind General set v2020.[7] Another example is the October 2023 AlphaFold release; though they reported an accuracy of 73.6% on the PoseBusters Benchmark, this accuracy dropped to 50% when test proteins and ligands were novel*.[8] While model memorization can be useful if your target of interest is from your training set, many targets are novel and ligand novelty is critical in the commercial success of drug development. For these reasons, we’ve used a train/test split with 30% protein sequence identity and 0.5 Tanimoto similarity (using 2048 bit RDKit fingerprints). We provide performance of our model on both total dataset and datasets filtered by similarity where possible.
First and foremost, BiosimDock dramatically outperforms other models on predicting small molecule binding affinities on the PDBbind core dataset (Figure 1). The results below illustrate the correlation between binding energy prediction and actual experimental binding affinity for BiosimDock, GNINA, and AutoDock Vina. For our model, BiosimDock, we report two results: one using the 30% sequence identity test-train split (BiosimDock Score), and one where we have removed only exact matches from our training set (BiosimDock Score*). For GNINA and Vina, we use the software provided by authors for benchmarking. A more accurate binding energy prediction translates to greater ability to rank molecules by binding affinity, improving the ability to distinguish true binders from false positives.
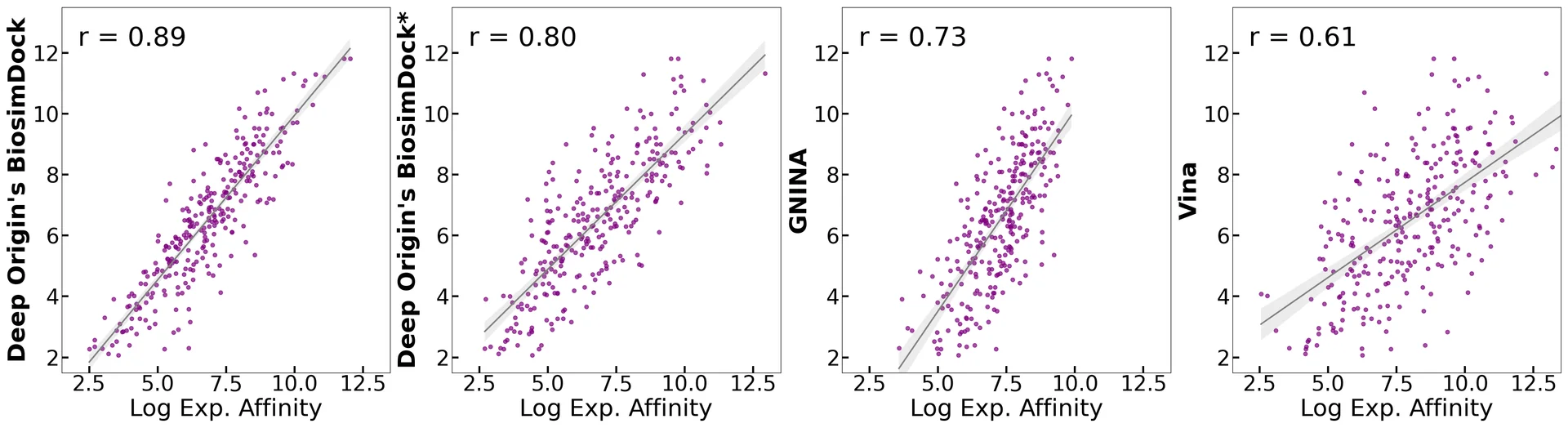
Figure 1. Correlation between the docking scores (absolute values) and log experimental binding affinity for Deep Origin BiosimDock, Deep Origin BiosimDock trained only on protein sequences with 30% or less homology and ligands with 0.5 or less Tanimoto similarity versus test set, GNINA, and Autodock Vina. The dataset is the PDBbind core set (285 protein-ligand complexes with measured dose-dependent experimental affinities).
Similarly, our model outperforms others in predicting the binding pose of ligands in the PDBbind core dataset. Figure 2 plots the percent of target-ligand pairs whose pose predictions in comparison to the actual crystal structure were within 2 Å-the accuracy cutoff typically considered ‘good’ for drug discovery. BiosimDock predicts 69% of PDBbind’s 285 structure-ligand complexes to be within 2 Å, compared to AutoDock Vina’s 50% and DOCK 6’s 38%. BiosimDock also outperforms DiffDock’s reported pose prediction accuracy of 39%.[9] This translates to better evaluation of docked molecules, including more accurate inputs to downstream molecular modeling like free energy of binding predictions.
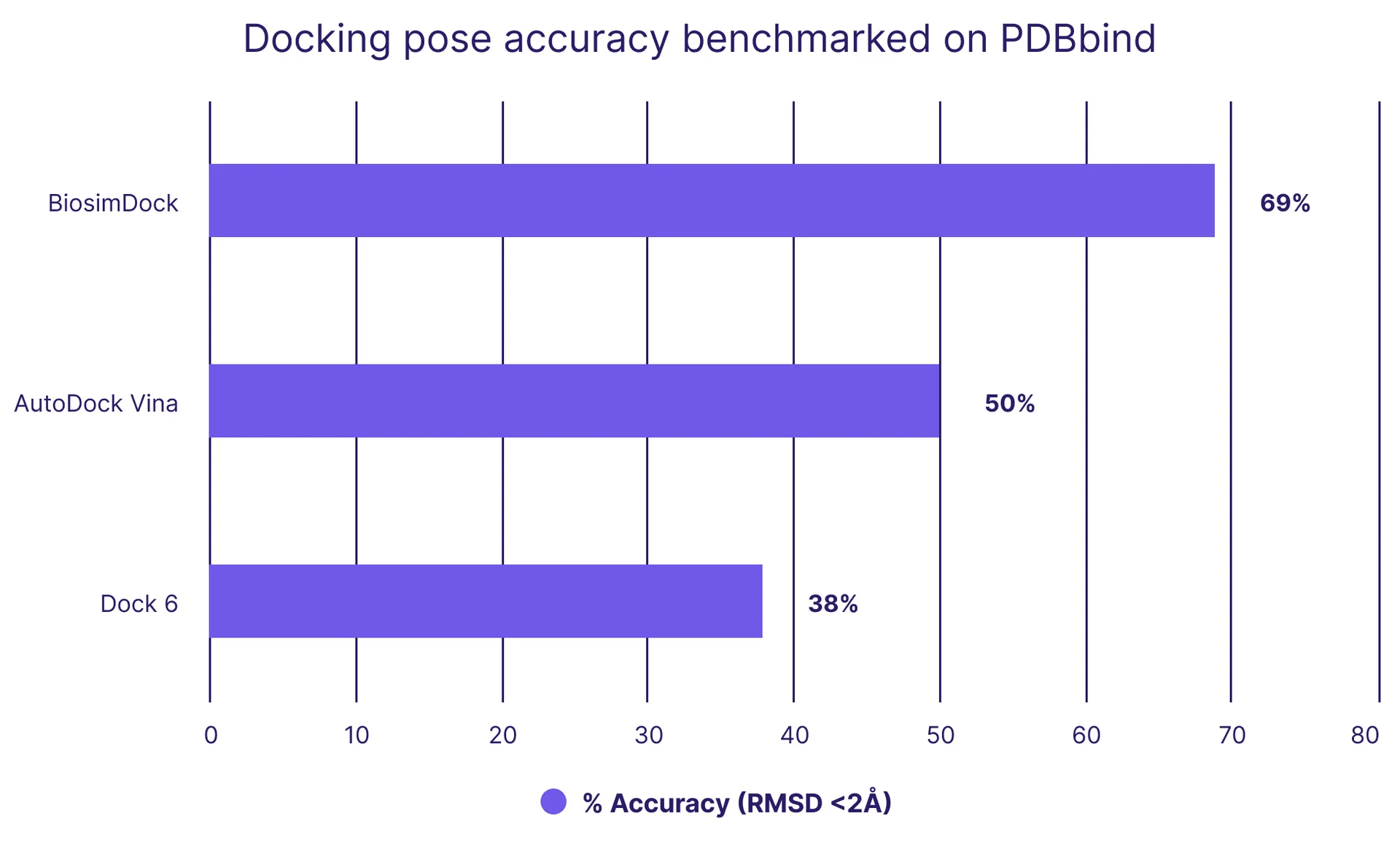
Figure 2. We outperform other models on docking accuracy, as benchmarked on the PDBbind core dataset, which contains crystal structures of 285 protein-ligand pairs. Accuracy is measured by the percentage of ligand poses predicted within 2 Å of experimental results, which is generally considered accurate for drug discovery. There are no error bars because each protein-ligand pair is assessed to have a binary ‘yes’ or ‘no’ value.
It is crucial to understand both the strengths and limitations of our models, so we analyzed BiosimDock’s performance on different target protein classes (Figure 3) using Expasy’s enzyme classifications**. Our analysis revealed a strong capability in accurately predicting ligand poses for receptor-ligand structures. But we observed comparatively lower performance in predicting ligand poses for lyases, including enzymes such as oxidoreductases, ribonucleases, and dehydratases. This means we aren’t stopping the development work here -we are refining and enhancing our models in these specific areas.
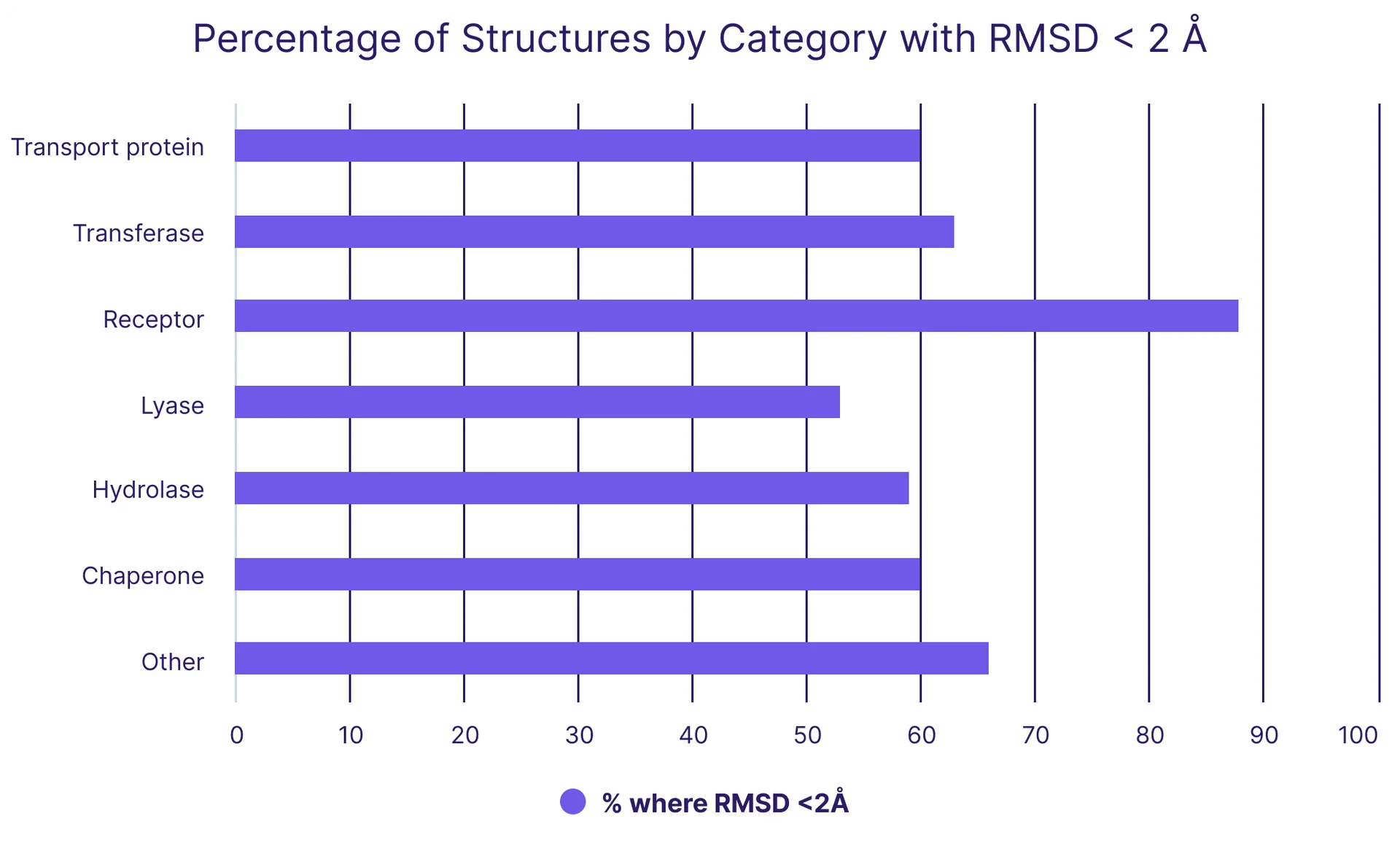
Figure 3. Our performance on predicting ligand binding poses by protein class, as defined by Expasy’s enzyme classifications. Accuracy is measured by the percentage of ligand poses predicted to be within 2 Å of experimental results, which is generally considered accurate for drug discovery. The dataset used is PDBbind core set; there are no error bars because each protein-ligand pair is assessed to have a binary ‘yes’ or ‘no’ value.
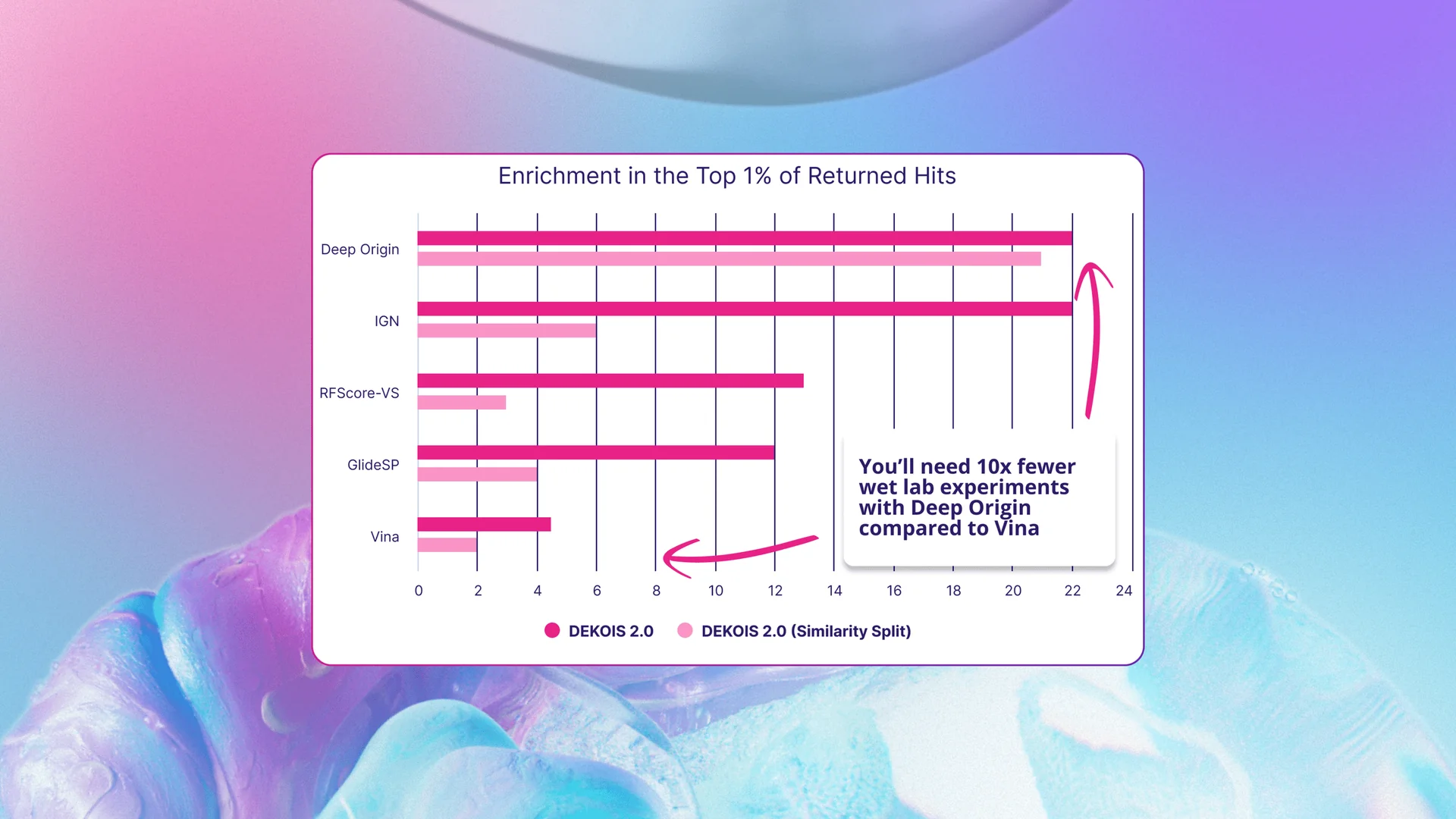
When benchmarked on DEKOIS 2.0, BiosimDock similarly outperformed compared to published data on other models, as measured by enrichment of true binders of false positives in the top 1% of ranked molecules (Figure 4). The published data on the performance of InteractionGraphNet (IGN), RFScore-VS, Schrödinger’s GlideSP, and AutoDock Vina enabled us to compare BiosimDock’s to these models.[10] We found that BiosimDock returned nearly double the number of true hits compared to RFScore-VS and GlideSP, and more than four times the number of true hits compared to Autodock Vina when assessed on all 81 DEKOIS 2.0 targets. The BiosimDock model performed similarly to IGN (dark pink bars in Figure 4).
But again, a common pitfall of AI models is exaggerated performance because test sets are contaminated with structures that are similar or identical to those in the training set. To address this, we removed targets similar to those in our training set and reassessed performance, comparing it to a similar exercise performed by IGN during benchmarking (light pink bar in Figure 4). While IGN’s performance dropped substantially when similar structures were filtered out of the test set, the BiosimDock model’s performance remained robust.
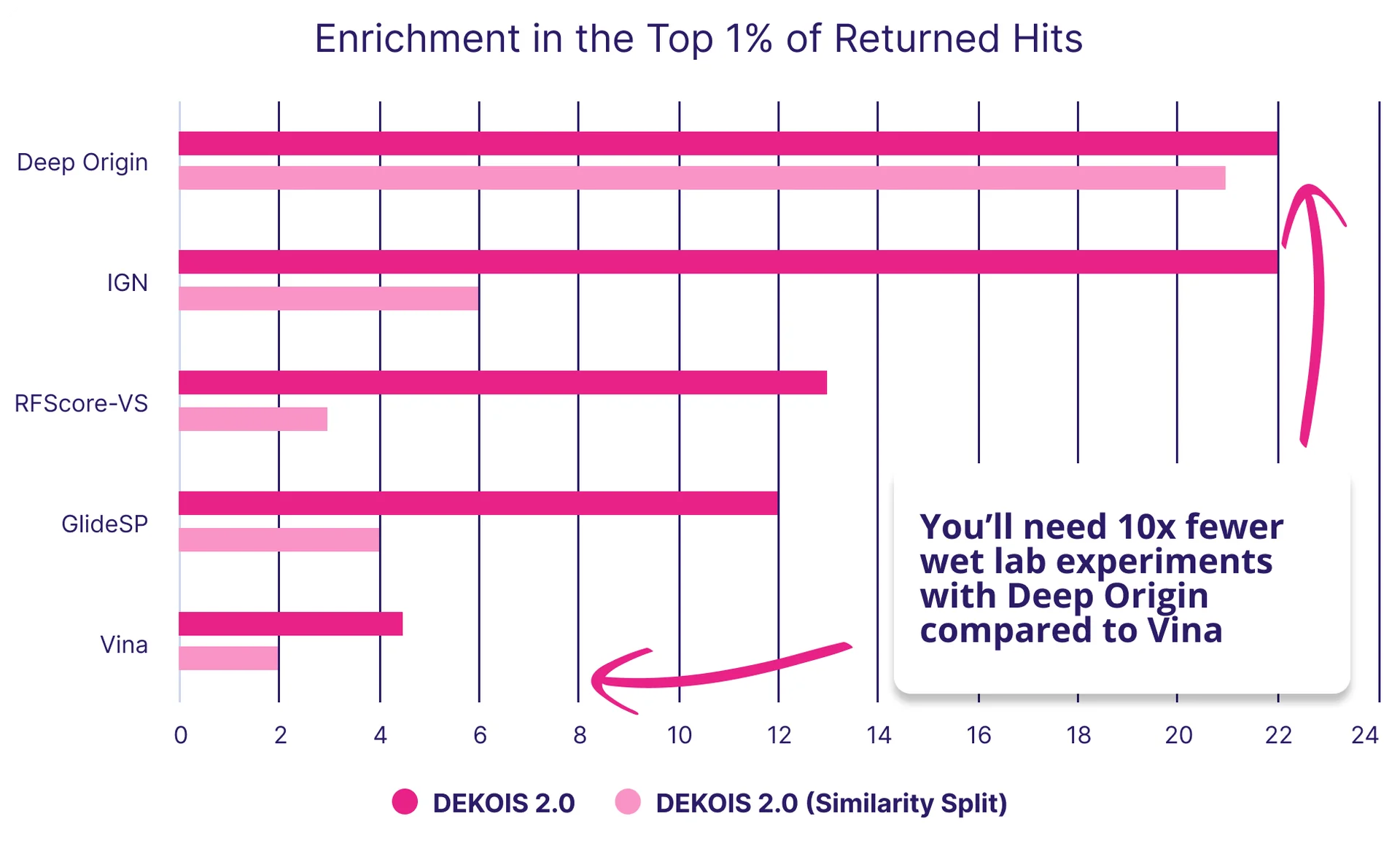
Figure 4: We outperform some popular docking tools, including industry standard such as GlideSP9, in the ability to identify true binders from decoys. The DEKOIS 2.0 dataset contains 80 target proteins with true binders and decoy molecules that are similar in physical and chemical properties, but do not bind the target protein. Dark pink bars are for all data, while light pink bars (Similarity Split) are the results after filtering out targets and ligands from the test data similar to training data.
We screen over 50 billion to find potential hits
That was a boatload of data we just threw at you. Let’s take a breather and talk about really big pools - of molecules, that is In our last blog post, we highlighted trends in the literature that suggest bigger screens lead to identification of more, better hits.[11], [12] Searching larger molecular spaces that haven’t already been searched over by others should also provide novel molecules for intellectual property claims.
Access to computational resources has grown exponentially, but it’s still worth noting that not all docking tools scale to enable virtual screening. In particular, not all of them are able to screen at scale. Nor are all tools able to deal with unenumerated spaces, which are combinations of molecular parts (synthons) and reactions, to put them together into molecules. Because of this, most virtual screens remain at the scale of 100 million molecules and are conducted on well-traversed libraries of molecules.
The cutting edge of virtual screening remains at the scale of 10s of billions of molecules (1010), though it’s possible that larger screens have been conducted and are yet unpublished. Today, state-of-the-art virtual screening technologies can be applied to routinely screen billion-compound libraries such as Enamine REAL Space, which is an unenumerated space of 48 billion molecules. Using BiosimVS, we can screen an unenumerated space of 50 billion molecules in a few days. The next frontier is the trillion molecule libraries, which we’re working on now. One challenge of working at this scale is that when the number of possible molecules increases, the number of false positives also increases.[11], [13] This is one of the reasons we have focused heavily on increasing our model’s ability to distinguish between true binders and false positives. If you’re interested in working with us to push the envelope and have a target of interest, feel free to drop us a line.
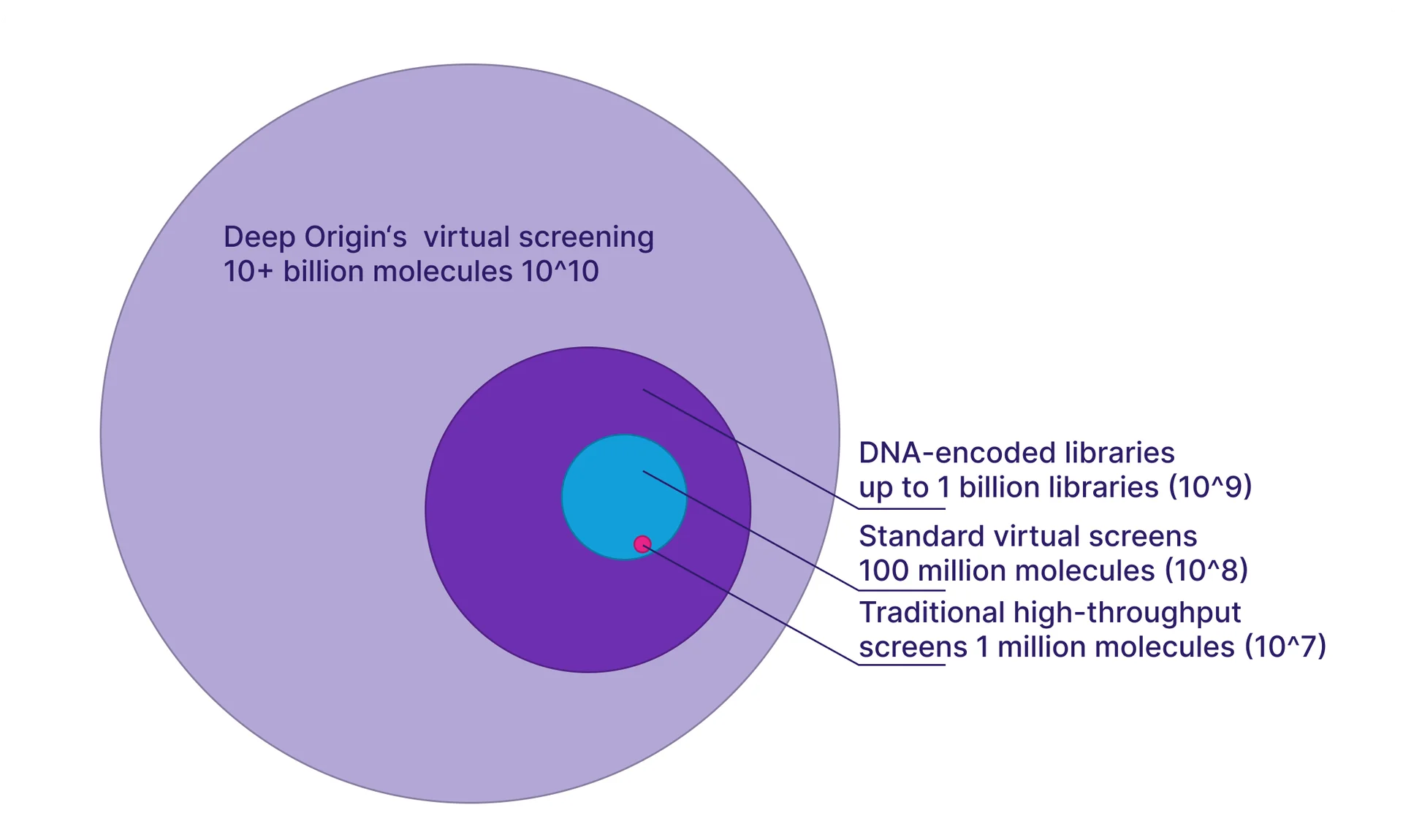
Figure 5. A roughly-to-scale illustration of searchable space with Deep Origin’s virtual screening, compared to DNA-encoded libraries, standard virtual screens, and traditional high-throughput screens.
We can optimize for multiple parameters at once
The ability of a molecule to bind to a target protein is just one property of a potential drug - and there are many more to optimize. Many of these properties impact the absorption, distribution, metabolism, excretion, and toxicology (ADMET) or formulation of a potential drug. While computational models exist to approximate many of these attributes, our exploration revealed deficiencies in both accuracy and accessibility of these models. Therefore, we are developing our own computational models to address these shortcomings.
While we’re still working on these property prediction models, below is a snapshot of our progress in predicting solubility properties such as logS, logP, and logD (Figure 6). We outperform many well-known models, as measured by Mean Average Error (MAE) or Root Mean Squared Error (RMSE). We are continuing to train these models and build additional models for hERG, toxicity and many other molecular property predictions.
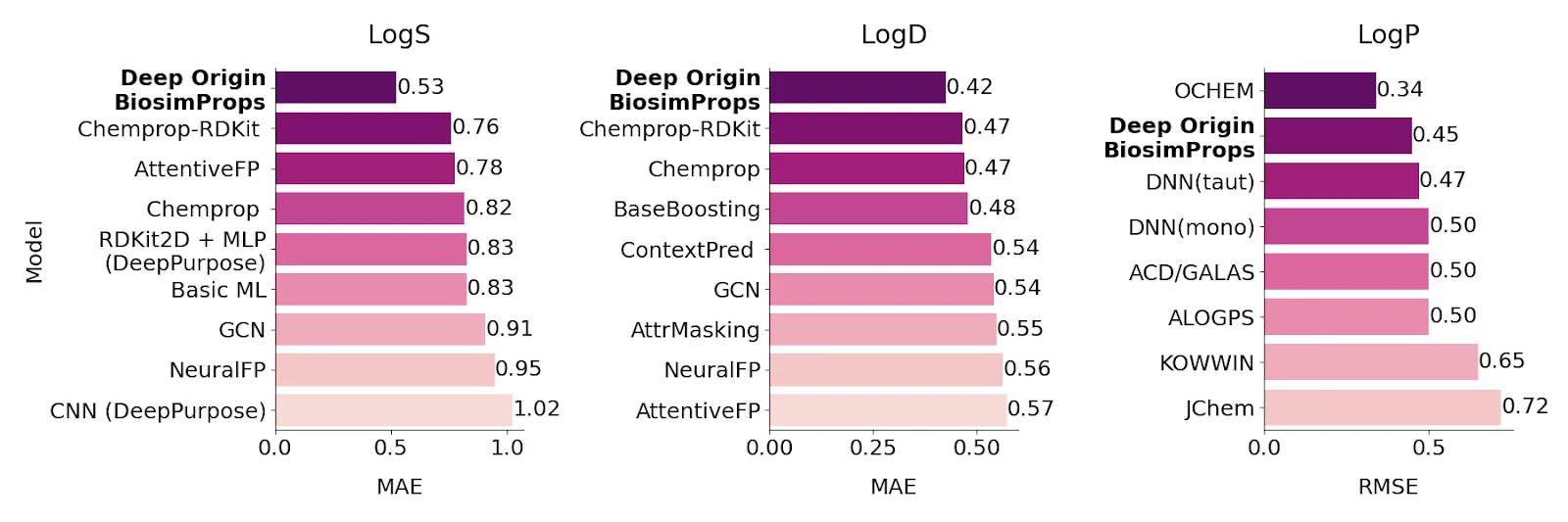
Figure 6.: Performance of our models for solubility in water (logS), simple octanol-water partition coefficient not accounting for molecular charge (logP), and a more realistic octanol-water partition coefficient accounting for pKa (logD). The performance is reported in mean absolute error (MAE) for logS and logD, and in root-mean-square error (RMSE) for logP, from Therapeutics Data Commons datasets. Lower numbers indicate less error, and thus greater accuracy.[14], [15], [16]
We rediscover known binders
While benchmarking is useful, it provides only perspective on the quality of the model. To test our tools in more realistic examples of drug discovery, we looked for examples where existing screening tools failed to identify drugs on challenging targets.
One example of this is the JAK2 pseudokinase (PK) domain. While it has fallen out of favor as a target, JAK PK is an interesting target because other commercial docking tools - Glide SP, Glide XP, and GOLD ChemScore - struggled to distinguish the 13 true binders from 30 false positive compounds.[17] Compared to the data in Cutrona et al., 2020, BiosimDock was able to better distinguish between true binders and false positives (Figure 7, left). We then tested the ability of BiosimVS and AutoDock Vina to identify these 13 true binders from a pool of 100,000 molecules with druglike properties, and found that BiosimVS was able to identify more true binders (Figure 7, right). This included filgotinib, an approved drug targeting JAK1 that is known to bind the JAK2 PK domain (Figure 8).[18] Interestingly, 10 true binders don’t rank in the top 100 - indicating there is more work for us to do.
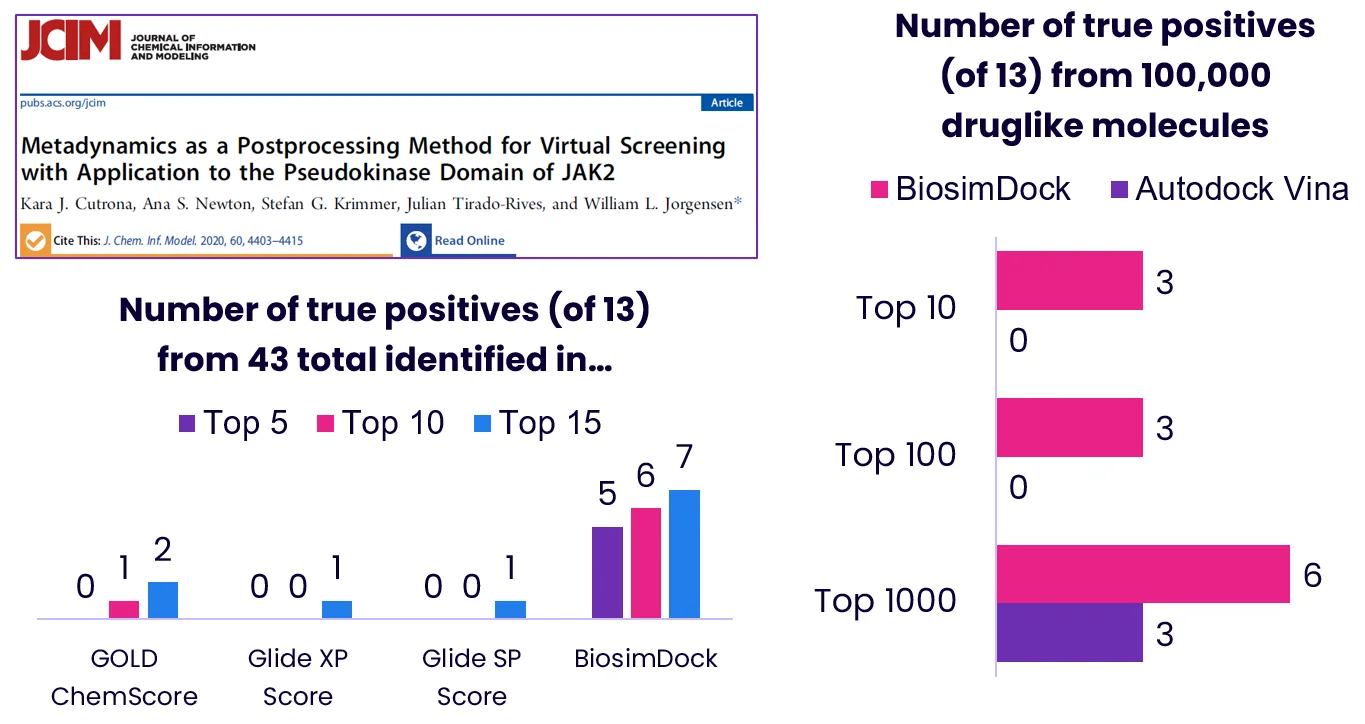
Figure 7.: Benchmarking our performance against well-known commercial docking software, using JAK2 pseudokinase as an example (screenshot of paper header).[17] We used the same PDB structure for JAK2 (PDB ID 4FVR) that is referenced in Cutrona et al., 2020.**
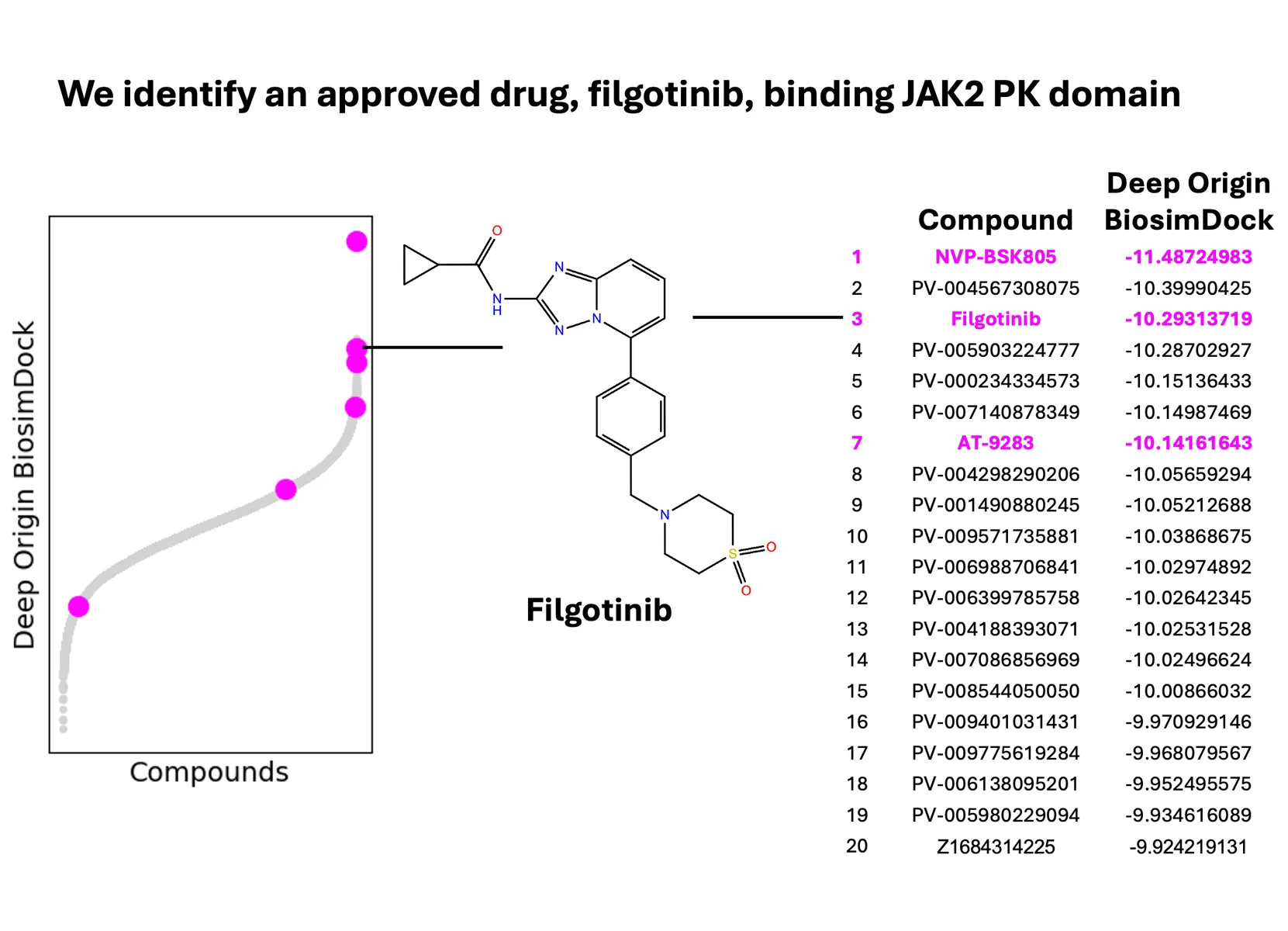
Figure 8.: We identify some known binders to the JAK2 pseudokinase (PK) domain.[18]
We also tested BiosimDock and BiosimVS on other challenging targets from different classes. For KRAS G12D, we tested BiosimVS and AutoDock Vina’s ability to prioritize 16 experimentally-validated binders from 100,000 molecules with druglike properties. We found that we were able to identify 13 of the 16 binders in the top 20 molecules, with Mirati Therapeutics’ MRTX1133 rising to the top (Figure 9). For protease DPP4, we tested BiosimVS’s ability to filter 2,830 experimentally-validated binders from 100,000 druglike molecules using docking with multiple conformations (Figure 10). We found that every molecule in the top 170 was a true binder, with no false positives. Moreover, analogs of approved drugs are among the molecules identified.
We’re continuing to validate our models on additional targets internally and in partnerships. If you’re interested, you can contact us here.
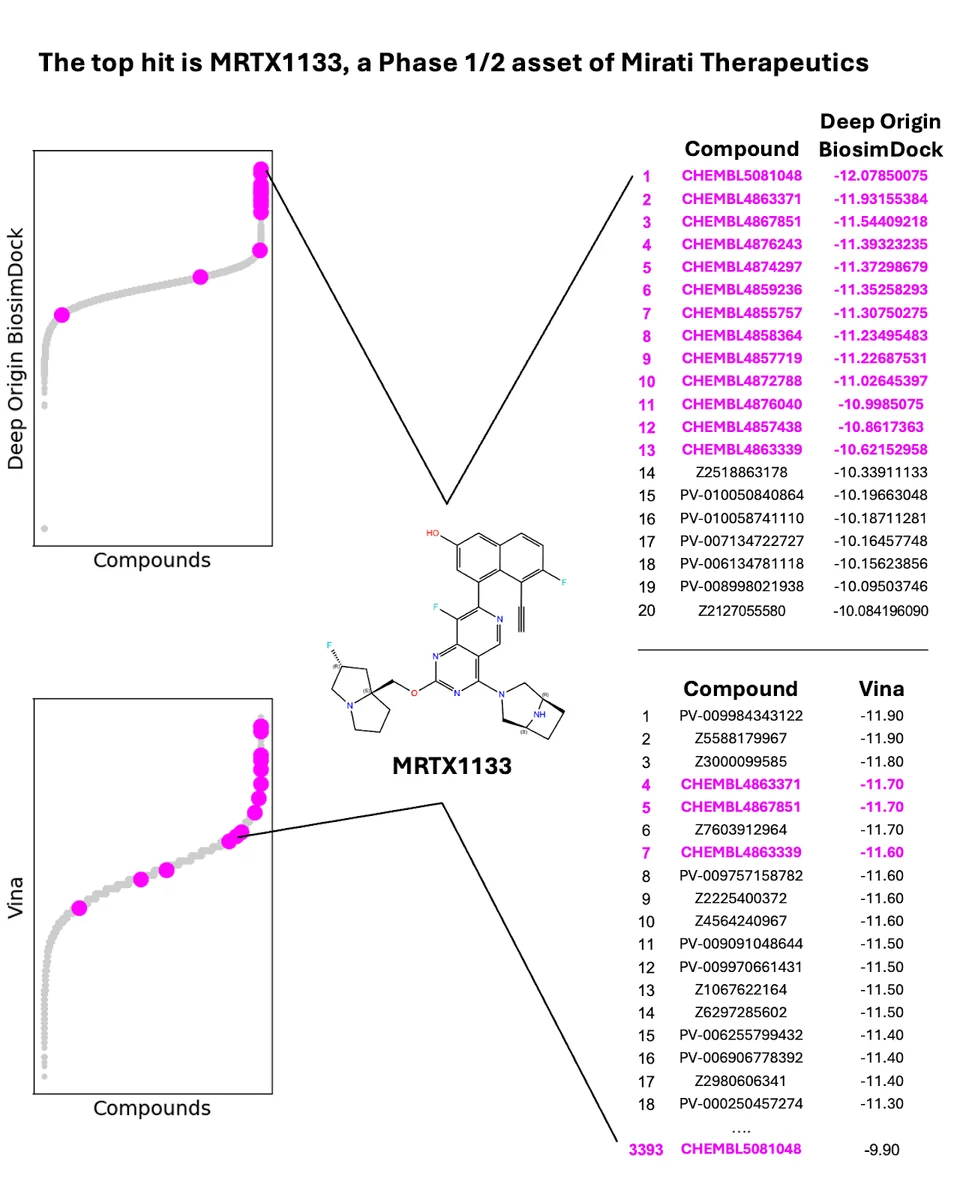
Figure 9. We re-discover known, experimentally-validated binders of KRAS G12D at a much higher rate than Autodock Vina. Starting library is 16 experimentally validated binders in a pool of 100,000 randomly selected druglike molecules; PDB ID 7RPZ used for docking.
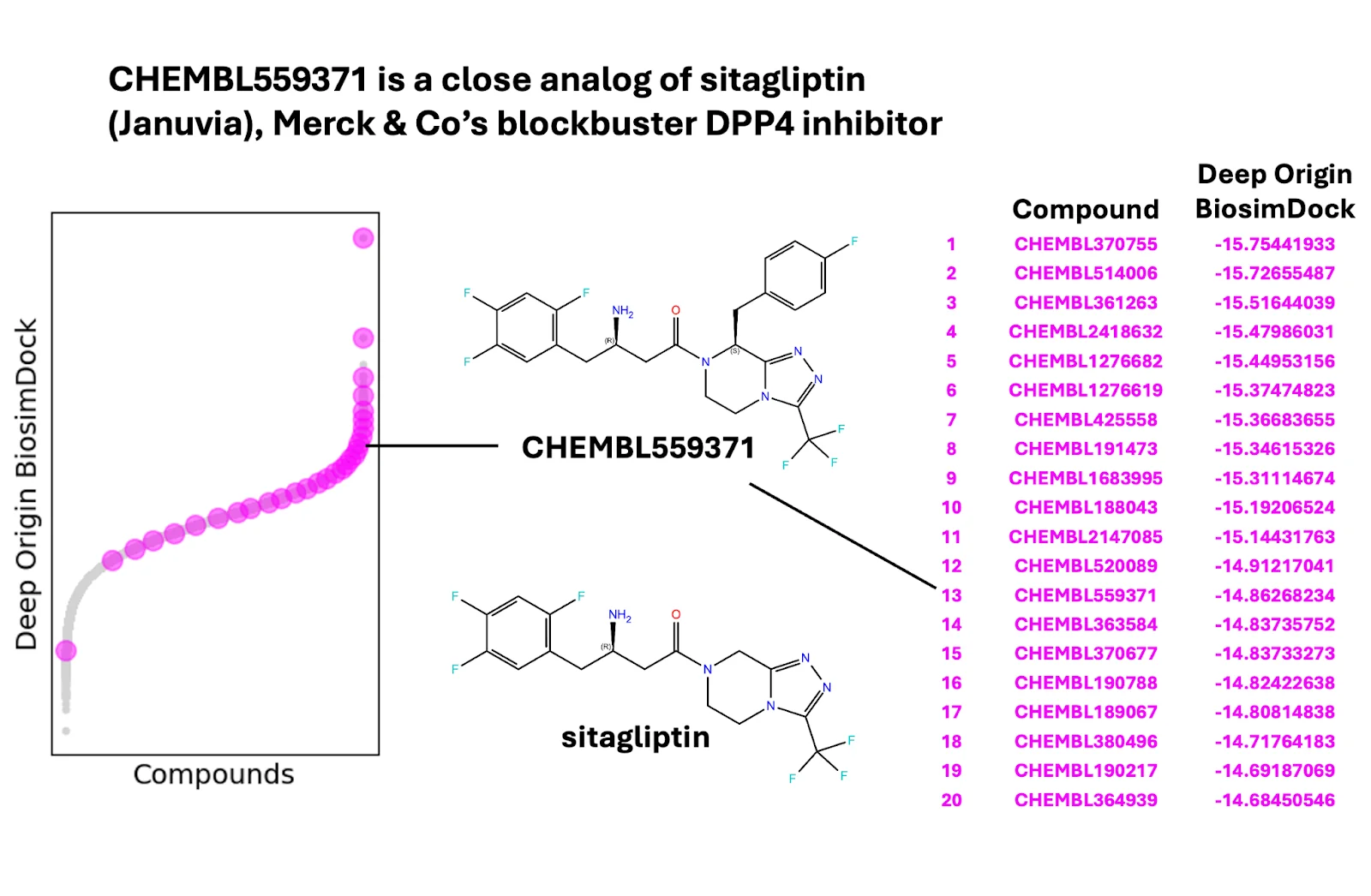
Figure 10. We rediscover known, experimentally-validated binders of DPP4, for an enrichment factor of 363-fold. Starting library is 2,830 experimentally validated binders in a pool of 100,000 randomly selected druglike molecules. All molecules in the top 170 are true binders. For clarity the figure shows 30 representative compounds of 2,830 after linear sampling. PDB IDs 1X70, 3G0B, 4A5S, and 6B1E were used for ensemble docking.
What we haven’t solved yet
We hope you enjoyed that whirlwind tour of what we’ve been building in drug discovery. While we’re excited about the results so far, there is still work to do and many unsolved problems.
You may have noticed that we did not address challenge #1, the need for a deep understanding of a target’s biology and structure. We can’t say much yet, but we’ll leave you with these two proteins hanging out…
Figure 11. A 1 µs all-atom molecular dynamics simulation of MDM2-p53 interaction. MDM2 (gray with electrostatic potential surface) is an E3 ligase for tumor suppressor p53 protein (p53 peptide is shown in cyan). Simulations started from PDB ID 4HFZ upon reverting MDM2 to wild type.
Want to try the models?
Though we can’t make all of our tools publicly available, you can try out BiosimDock and some BiosimProps models today as part of a limited beta access launch of Balto, our AI Assistant in Drug Discovery. You can sign up here.
Happy drughunting.
Footnotes:
* In the October 2023 AlphaFold study, a protein was considered novel based on a 40% template identity cut off, and a ligand based on a 0.5 Tanimoto similarity coefficient cut off relative to train (using 2048 bit RDKit fingerprints).
** Expasy’s enzyme classification system applies only to enzymes, so we applied it to proteins with catalytic activity. Non-catalytic enzymes were classed as either ‘receptors’ or ‘other’. Enzymatic classes that contained fewer than 10 representatives in the PDBbind dataset were also classed as ‘other’.
References:
- http://www.pdbbind.org.cn/
- https://www.pharmchem.uni-tuebingen.de/dekois/
- Su, M., Yang, Q., Du, Y., Feng, G., Liu, Z., Li, Y., Wang, R., 2019. Comparative Assessment of Scoring Functions: The CASF-2016 Update. J. Chem. Inf. Model. 59, 895–913. https://doi.org/10.1021/acs.jcim.8b00545
- Bauer, M.R., Ibrahim, T.M., Vogel, S.M., Boeckler, F.M., 2013. Evaluation and Optimization of Virtual Screening Workflows with DEKOIS 2.0 – A Public Library of Challenging Docking Benchmark Sets. J. Chem. Inf. Model. 53, 1447–1462. https://doi.org/10.1021/ci400115b
- Ibrahim, T.M., Bauer, M.R., Boeckler, F.M., 2015. Applying DEKOIS 2.0 in structure-based virtual screening to probe the impact of preparation procedures and score normalization. J Cheminform 7, 21. https://doi.org/10.1186/s13321-015-0074-6
- Boeckler, F.M., Bauer, M.R., Ibrahim, T.M., Vogel, S.M., 2014. Use of DEKOIS 2.0 to gain insights for virtual screening. J Cheminform 6, O24. https://doi.org/10.1186/1758-2946-6-S1-O24
- Buttenschoen, M., Morris, G.M., Deane, C.M., 2023. PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences.
- https://storage.googleapis.com/deepmind-media/DeepMind.com/Blog/a-glimpse-of-the-next-generation-of-alphafold/alphafold_latest_oct2023.pdf
- Corso, G., Stärk, H., Jing, B., Barzilay, R., Jaakkola, T., 2022. DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking. https://doi.org/10.48550/ARXIV.2210.01776
- Jiang, D., Hsieh, C.-Y., Wu, Z., Kang, Y., Wang, J., Wang, E., Liao, B., Shen, C., Xu, L., Wu, J., Cao, D., Hou, T., 2021. InteractionGraphNet: A Novel and Efficient Deep Graph Representation Learning Framework for Accurate Protein–Ligand Interaction Predictions. J. Med. Chem. 64, 18209–18232. https://doi.org/10.1021/acs.jmedchem.1c01830
- Lyu, J., Irwin, J.J., Shoichet, B.K., 2023. Modeling the expansion of virtual screening libraries. Nat Chem Biol 19, 712–718. https://doi.org/10.1038/s41589-022-01234-w
- Sadybekov, A.V., Katritch, V., 2023. Computational approaches streamlining drug discovery. Nature 616, 673–685. https://doi.org/10.1038/s41586-023-05905-z
- https://www.science.org/content/blog-post/screening-big-libraries-how-s-it-going
- TDC.Solubility_AqSolDB
- Ulrich, N., Goss, K.-U., Ebert, A., 2021. Exploring the octanol–water partition coefficient dataset using deep learning techniques and data augmentation. Commun Chem 4, 90. https://doi.org/10.1038/s42004-021-00528-9
- TDC.Lipophilicity_AstraZeneca
- Cutrona, K.J., Newton, A.S., Krimmer, S.G., Tirado-Rives, J., Jorgensen, W.L., 2020. Metadynamics as a Postprocessing Method for Virtual Screening with Application to the Pseudokinase Domain of JAK2. J. Chem. Inf. Model. 60, 4403–4415. https://doi.org/10.1021/acs.jcim.0c00276
- Newton, A.S., Deiana, L., Puleo, D.E., Cisneros, J.A., Cutrona, K.J., Schlessinger, J., Jorgensen, W.L., 2017. JAK2 JH2 Fluorescence Polarization Assay and Crystal Structures for Complexes with Three Small Molecules. ACS Med. Chem. Lett. 8, 614–617. https://doi.org/10.1021/acsmedchemlett.7b00154