Your Whole Drug Discovery Workflow in a Single API
Instant API access to our state-of-the-art molecular and cheminformatics models. Build your own drug discovery workflows, no configuration or DevOps needed.
# Find binding pockets
protein, pockets = pocket_finder_client.find_pockets(
protein, num_pockets=10, pocket_min_size=30)
# Dock ligands to protein
report = docking_client.dock(
protein=protein,
ligands=ligands,
pocket_data=pocket_data
)
# Get ADMET predictions
for result in report.results:
result.top_ligand.admet_properties()Powerful Capabilities, Simple Interface
Everything you need for computational drug discovery, accessible through clean Python APIs
Pocket Finding & Analysis
Identify and compare binding pocket quality with minimal code. Features include pocket creation from crystal structures, novelty assessment, and coordinate/residue ID filtering.
protein, pockets_report = pocket_finder_client.find_pockets(
protein, num_pockets=10, pocket_min_size=30)| Pocket Id | Color | Druggability Score | Volume | Total S A S A |
|---|---|---|---|---|
| 1 | Red | 0.824 | 393.000 | 347.516 |
| 2 | Green | 0.824 | 1692.000 | 919.499 |
| 3 | Blue | 0.655 | 520.000 | 492.714 |
Docking Grid Visualization
Validate pocket boundaries and grid box dimensions using precomputed or custom protein data.
pocket_data.show_box()| Parameter | Value |
|---|---|
| Center | (12.45, -8.22, 24.88) |
| Size | 22 × 22 × 22 Å |
Docking Scores & Reporting
Execute docking with single function calls, generating poses, scores, and interactive visualizations within seconds.
report = docking_client.dock(
protein=protein, ligands=ligands, pocket_data=pocket_data,
protinoate_ligands=False,
)| Smiles | Ranking Score | Binding Energy |
|---|---|---|
| CC(C)Cc1ccc... | 48.139 | -7.063 |
| Cn1c(=O)c2c... | 48.073 | -5.230 |
| CC(=O)Oc1ccccc1... | 47.530 | -5.526 |
ADMET Predictions
Predict absorption, distribution, metabolism, excretion, and toxicity properties for seamless pipeline integration.
for result in report.results:
result.top_ligand.admet_properties()| Property | Value | Status |
|---|---|---|
| LogP | 0.46 | Optimal |
| Solubility | -1.8 | Good |
| hERG Risk | Low | Safe |
| CYP450 2D6 | Non-inhibitor | Safe |
50+ Tools at Your Fingertips
A comprehensive toolkit for every stage of drug discovery
Molecular Data Retrieval
Protein & Ligand Processing
Molecular Dynamics
Molecular Properties
Visualization
Data Processing
Backed by Research
Our API is built on peer-reviewed science and validated methodologies
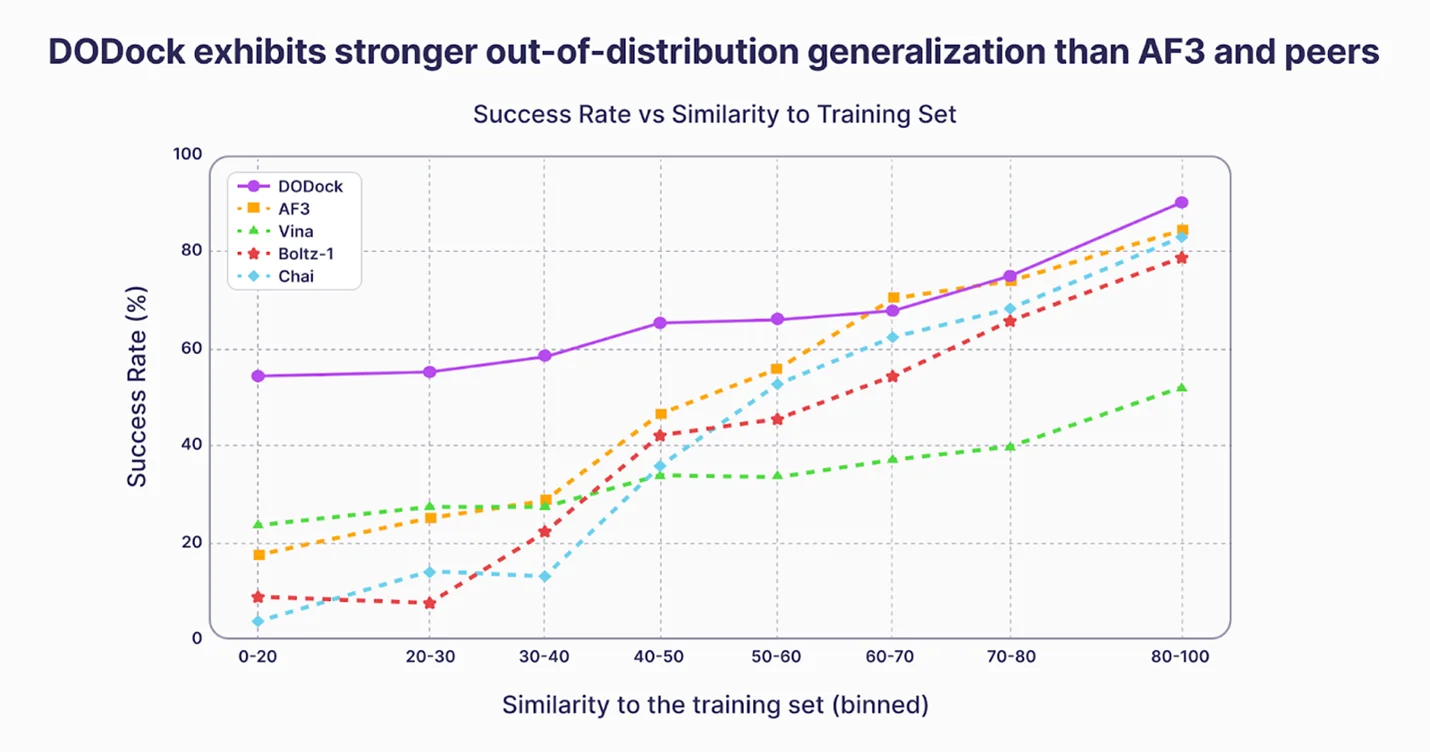
Deep Origin Congratulates Isomorphic Labs on Catching Up
After presenting comparable docking results last year—and using them to prospectively identify drug candidates on difficult targets—Deep Origin welcomes Isomorphic Labs to the frontier.
Read articleAccelerating Drug Discovery with Physics-Informed Machine Learning
Addressing Drug Discovery Challenges with a Multiscale Molecular Modeling Pipeline
Discovering Novel Synthetic Lethal Pairs With Large Scale Cellular Simulations
Finding Promising Molecules and Biological Background With Balto
Out-of-the-Box GenAI for Medicinal Chemistry
Get Started
Request access to start building with our drug discovery API.
No spam, ever. We'll only email you about API access.