In the world of drug development, inefficiencies and inaccurate tools at the earliest stages of discovery can cost millions of dollars and years of time. Any time and effort that teams can save early on is big. It’s in this context that we’re thrilled to say that we have something to help. And we want you to be the first to benefit from it.
Here’s what we’ve been working on:
We built one of the most performant molecular modeling algorithms available (beating out common tools like AutoDock Vina and Schrodinger’s Glide with 10-fold better enrichment factors). But as we all know, drug discovery is hard. Even 10x the needles in a haystack are hard to find.
How do you magnify this 10x improvement by even more? Make your tool accessible to a wider audience. In biotechs and pharma, that means making these tools accessible to medicinal chemists.
In many settings medicinal chemists outnumber computational chemists 10 to
- The back and forth in designing molecules and assaying them in the lab to test hypotheses is a major time suck in the early stages of drug discovery. Though they have access to some computational tools like Chemd3D, historically they’ve been shut out of more predictive, cutting-edge simulation tools because they require months of training (and cost $100,000’s a year in licensing fees).
This is where Balto comes in. Balto is a simple conversational interface that lets you quickly gather and manipulate chemical data from across the web and immediately apply powerful, industry-leading molecular simulations. Our custom-trained chem-aware AI helps medicinal chemists get up-to-speed rapidly, offering suggestions for how to perform analysis and the ability to run a wide array of computational algorithms behind the scenes.
Want to try out Balto - our AI assistant for molecular modeling? Get one month free here.
Molecular Docking, No Special Training Required
In traditional setups, molecular docking is the domain of computational chemists, with complex tools that require months of training. Balto turns this on its head. With Balto’s AI-driven interface, medicinal chemists can now perform precise docking simulations just by asking. Whether it’s testing a hypothesis or exploring a new target, Balto’s docking capabilities give you sophisticated, real-time insights without the need for a specialist.
By making advanced docking accessible, Balto frees up resources, allowing medicinal chemists to test more ideas quickly and refine compounds before any lab work begins. This is how Balto accelerates the discovery pipeline, making high-quality predictions available to the people who need them most.
Under the hood, Balto is powered by our own docking model, which outperforms DiffDock, AutoDock Vina, and Schrodinger’s Glide on enriching for true binders. Which means more true positives out of docking, and less chance of stumbling down the wrong path.
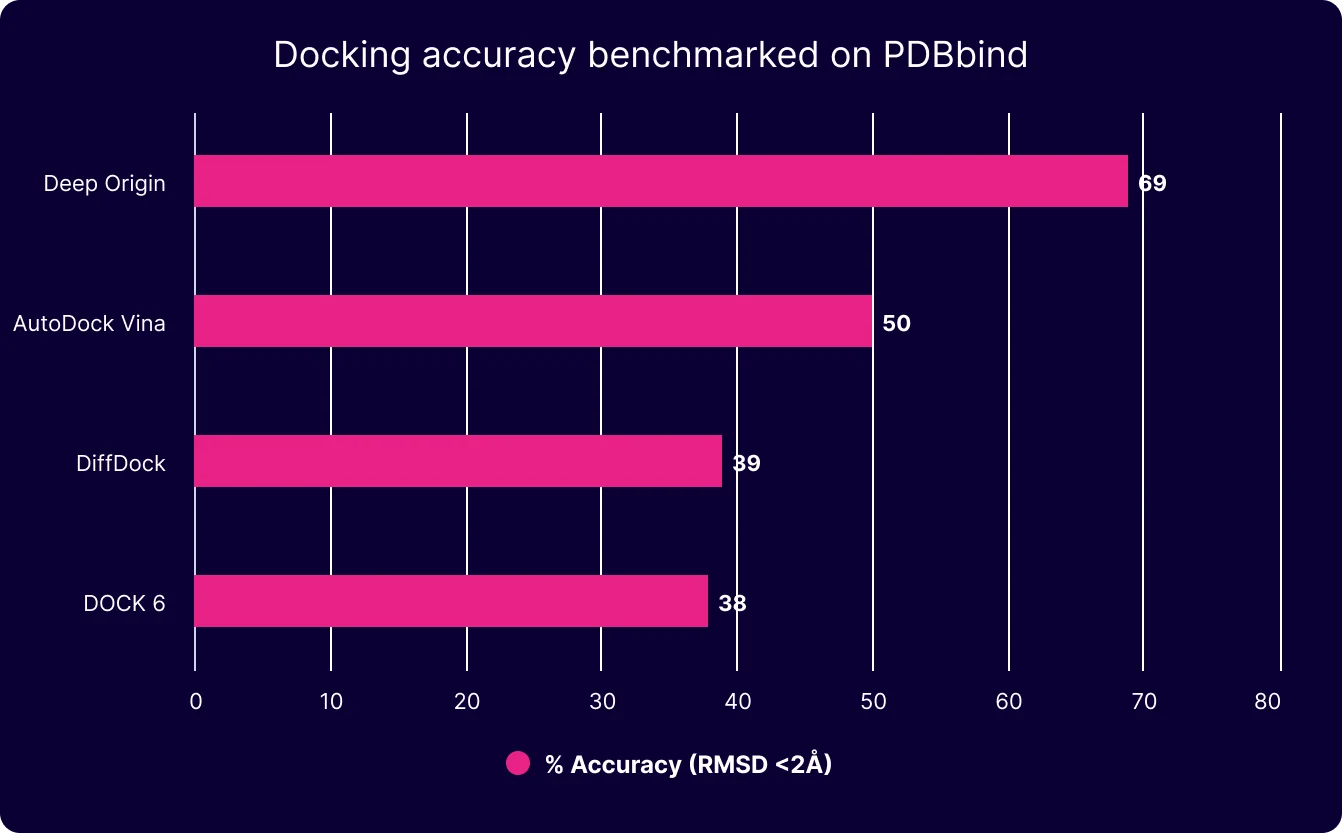
Benchmarked on the PDBBind 285 set (1)
Discover Key Binding Sites with Ease
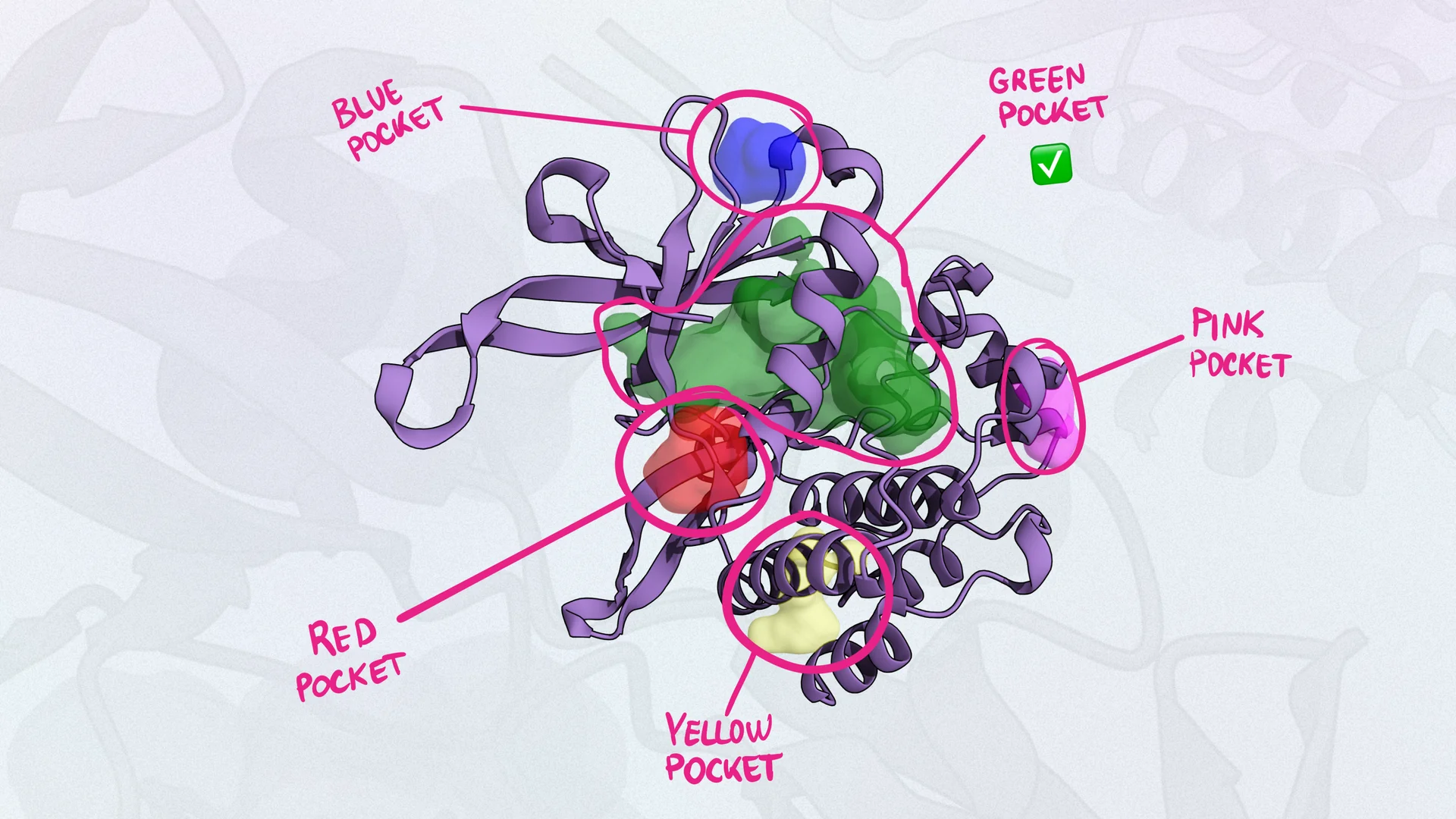
Identifying the perfect binding pocket is crucial for drug design, but pinpointing it can be challenging and time-intensive. With Balto’s AI-powered pocket-finding tool, medicinal chemists can quickly identify and analyze binding sites without specialized training. Balto’s pocket-finding capabilities provide not just common crystal ligand binding sites but are also capable of revealing novel pockets that might otherwise require extensive computational expertise.
Balto’s user-friendly pocket-finding feature not only saves time but also ensures researchers have essential insights to move forward with docking.
A World of Chem Data You Can Do More Than Chat With
Balto goes beyond simple conversation. Unlike generic chat tools, Balto lets you load molecules, and pull data from PDFs—all in a seamless flow from information-gathering to active discovery. Medicinal chemists can now integrate academic literature and chemical data directly into Balto, where it becomes usable in real time for docking simulations and molecular analysis. It’s not just a chat; it’s an interactive workspace designed to move drug discovery forward with unprecedented speed.
Have five minutes? Why not catch up on the latest scientific literature with an AI assistant, on us.
Molecular Modeling Priced Democratically
Most commercial molecular modeling tools often come with a steep price tag, locking many researchers out with thousands per seat or even hundreds of thousands annually. Balto changes that. For just $32 a month, scientists get full access to Balto’s advanced capabilities, with a pay-per-action model that lets you control costs based on actual usage. For example, docking 100 molecules against a binding pocket costs $19. That’s it.
Balto is designed to democratize access to powerful AI tools, ensuring that any researcher, lab, or institution can leverage top-tier molecular modeling without breaking the bank.
Find more details on Balto pricing and features here.