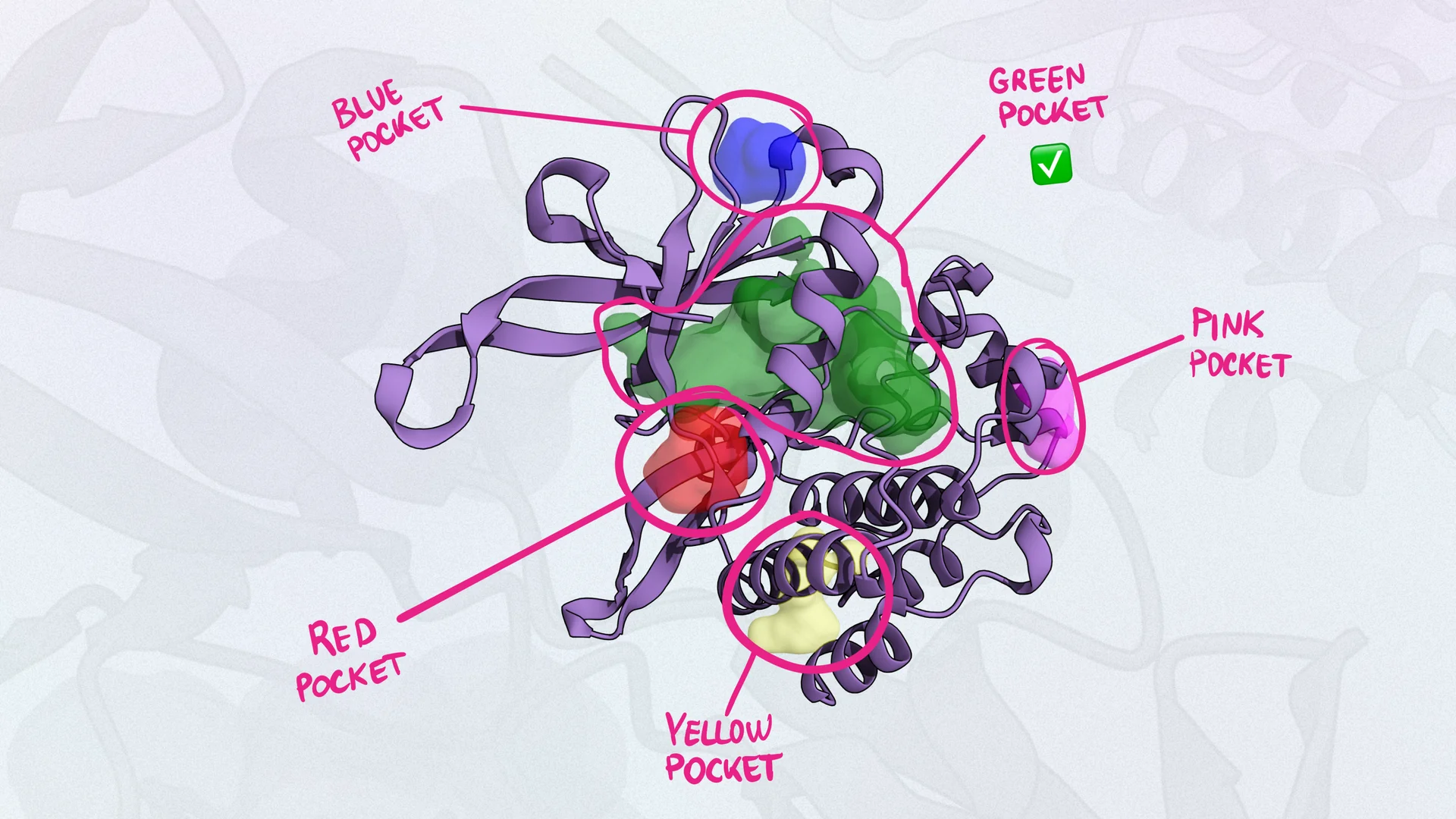
Density Functional Theory (DFT)
DFT provides detailed information about the electronic structure of drug molecules and their targets, which is crucial for understanding molecular interactions. It helps in elucidating reaction mechanisms by calculating transition states and energy barriers, aiding in the design of more efficient drugs.
Density Functional Theory (DFT) is a quantum mechanical method used to investigate the electronic structure of atoms, molecules, and condensed phases. DFT approximates the electron density of a system rather than the wave function, making it computationally more efficient while still providing accurate results for many systems. It is widely used in computational chemistry to study the properties of molecules, including their electronic structure, reactivity, and interaction with other molecules.
Importance in Computational Drug Discovery
- Electronic Structure Analysis: DFT provides detailed information about the electronic structure of drug molecules and their targets, which is crucial for understanding molecular interactions.
- Reaction Mechanisms: It helps in elucidating reaction mechanisms by calculating transition states and energy barriers, aiding in the design of more efficient drugs.
- Predicting Molecular Properties: DFT can predict various molecular properties such as dipole moments, polarizabilities, and redox potentials, which are important for ligand binding and drug design.
- Binding Affinity: By calculating the interaction energies between drug molecules and their targets, DFT can help predict binding affinities and optimize lead compounds.
- Molecular Dynamics Simulations: DFT can be integrated with molecular dynamics simulations to study the dynamic behavior of drug-target complexes at an atomic level.
Key Tools
- Gaussian: A widely used software package for performing DFT calculations, providing a range of functionalities for electronic structure analysis.
- ORCA: An efficient and flexible DFT software package suitable for studying the electronic structure of large molecules.
- VASP (Vienna Ab initio Simulation Package): A powerful tool for performing DFT calculations on periodic systems, widely used in material science and solid-state physics.
- Quantum ESPRESSO: An open-source suite for electronic-structure calculations and materials modeling, based on DFT.
- ADF (Amsterdam Density Functional): A DFT software package tailored for studying molecules and molecular properties.
Literature
“In vitro antimicrobial activities, molecular docking and density functional theory (DFT) evaluation of natural product-based vanillin derivatives featuring halogenated azo dyes.”
- Publication Date: 2023-09-26
- DOI: 10.1080/14786419.2023.2262713
- Summary: This study introduces diazo coupling to generate vanillin derivatives with effective antimicrobial properties. DFT calculations using Gaussian 09 supported the potential of vanillin-azo as an antimicrobial drug by analyzing binding affinity and energy gaps.
“Molecular Docking, Drug-Likeness and ADMET Analysis, Application of Density Functional Theory (DFT) and Molecular Dynamics (MD) Simulation to the Phytochemicals from Withania Somnifera as a Potential Antagonist of Estrogen Receptor Alpha (ER-α).”
- Publication Date: 2020-07-30
- DOI: 10.2174/1573409916999200730181611
- Summary: Investigates phytochemicals from Withania somnifera as potential ER-α antagonists. DFT was used to check the reactivity of selected molecules and their stability was confirmed using molecular dynamics simulations.
“Quantum Chemical Calculations on Locked Nucleic Acid based Antisense Modifications: A Density Functional Theory (DFT) Study at Monomer Level”
- Publication Date: 2020-07-16
- DOI: 10.26434/chemrxiv.12661628
- Summary: Uses DFT to investigate the quantum chemical parameters of antisense modifications. The study proposes novel LNA-based modifications and analyzes their reactivity and stability.
“Understanding the Structure–Activity Relationship through Density Functional Theory: A Simple Method Predicts Relative Binding Free Energies of Metalloenzyme Fragment-like Inhibitors”
- Publication Date: 2023-06-06
- DOI: 10.1021/acsomega.2c08156
- Summary: Applies DFT to predict binding free energies and understand the structure-activity relationship of metalloenzyme inhibitors, highlighting the electronic properties that differentiate strong and weak inhibitors.
“DFT‐Guided Discovery of Ethynyl‐Triazolyl‐Phosphinates as Modular Electrophiles for Chemoselective Cysteine Bioconjugation and Profiling”
- Publication Date: 2022-07-06
- DOI: 10.1002/anie.202205348
- Summary: Reports DFT-guided discovery of ETPs for cysteine-selective bioconjugation, demonstrating their potential in protein-protein conjugation and mass spectrometry-based cysteine profiling.